Meet Mitoxyperiosis: Cell's Newly Defined Mitochondrial Death Program
Cell death is a core physiological process in life activities, and its abnormal regulation is closely related to various diseases such as inflammation, tumors, and neurodegenerative diseases. Currently known modes of cell death include apoptosis, pyroptosis, necroptosis, and ferroptosis, but in the complex pathophysiological microenvironment of the body, are there any undiscovered novel modes of cell death? Recently, Wang et al. published a study entitled "Innate immune and metabolic signals induce mitochondria-dependent membrane lysis via mitoxyperiosis", which for the first time reported a novel mode of cell death-mitoxyperiosis (mitochondria-mediated cell lysis)-induced by the synergistic action of innate immune activation and metabolic disorders.
Innate immune activation (such as TLR ligand stimulation) and changes in the metabolic microenvironment (such as nutrient deprivation) are common physiological states when the body responses to infection and injury. While their individual effects on cell survival are limited, it remains unclear whether their synergistic action triggers a specific cell death program. Previous studies have shown that innate immune activation can induce the release of pro-inflammatory cytokines, and metabolic disorders can disrupt cellular homeostasis. It is hypothesized that these two factors may regulate cell death processes through synergistic effects. Based on this, the team established an "innate immune activation + metabolic disorder" (IIAMD) research model, using lipopolysaccharide (LPS, a TLR4 ligand) combined with carbon starvation (CS) as the core stimulating conditions, to systematically explore the characteristics and mechanisms of cell death induced by this combination.
IIAMD Synergistically Induces Inflammatory Cell Death in Macrophages
The team first verified the effect of IIAMD on macrophage survival, with the following core results:
-
LPS and CS synergistically induce cell death: Neither LPS, CS, nor other TLR ligands (such as PAM3, R837) alone could effectively induce death in bone marrow-derived macrophages (BMDMs), while IIAMD combinations such as LPS+CS, PAM3+CS, and R837+CS significantly induced cell death, as evidenced by increased lactate dehydrogenase (LDH) release and increased PI staining positivity;
-
IIAMD activates the NLRP3 inflammasome: LPS+CS treatment induced NLRP3 inflammasome activation, manifested as ASC speck formation and caspase-1 cleavage activation (P45→P20), and promoted the release of pro-inflammatory cytokines such as IL-1β and IL-18;
-
Signaling pathway depends on TLR/TNFR-MyD88: Cell death induced by TNF+CS depends on TNF receptors (Tnfrsf1a/Tnfrsf1b), while death induced by TLR ligands such as LPS and PAM3 + CS depends on the TLR downstream adaptor MyD88. MyD88 knockout cells were significantly resistant to IIAMD-induced death, while TNF knockout did not affect the lethal effect of TLR ligand + CS.
Glutathione Depletion and Oxidative Stress are the Core Driving Forces of IIAMD-Induced Cell Death
To clarify the core regulatory factors of the novel cell death induced by IIAMD, the team conducted non-targeted metabolomics analysis and found:
-
IIAMD significantly disrupts glutathione (GSH) metabolism: After LPS+CS treatment, 209 out of 231 differentially expressed metabolites in the cells showed decreased levels, with GSH metabolic pathways being significantly enriched and GSH levels significantly reduced;
-
Oxidative stress is critical for cell death: fluorescent probe detection showed that intracellular oxidative stress levels gradually increased after LPS+CS treatment, and all dying cells (PI-positive) were accompanied by the formation of oxidative stress foci; exogenous supplementation of GSH or the antioxidant N-acetylcysteine (NAC) dose-dependently inhibited cell death, proving that oxidative stress is a critical factor in lethality;
-
Oxidative stress depends on upstream TLR/TNFR signaling: LPS+CS-induced oxidative stress depends on the TLR4-MyD88 pathway, and TNF+CS-induced oxidative stress depends on TNFR signaling, consistent with the previously described cell death signaling pathway;
-
Distinct from oxidative stress-related oxeiptosis: Knocking down the core oxeiptosis molecule KEAP1 failed to inhibit IIAMD-induced cell death, and RNA-seq showed that NRF2 target gene expression was upregulated after LPS+CS treatment, contrary to the NRF2 inhibition characteristic of oxeiptosis, ruling out the possibility of it being oxeiptosis.
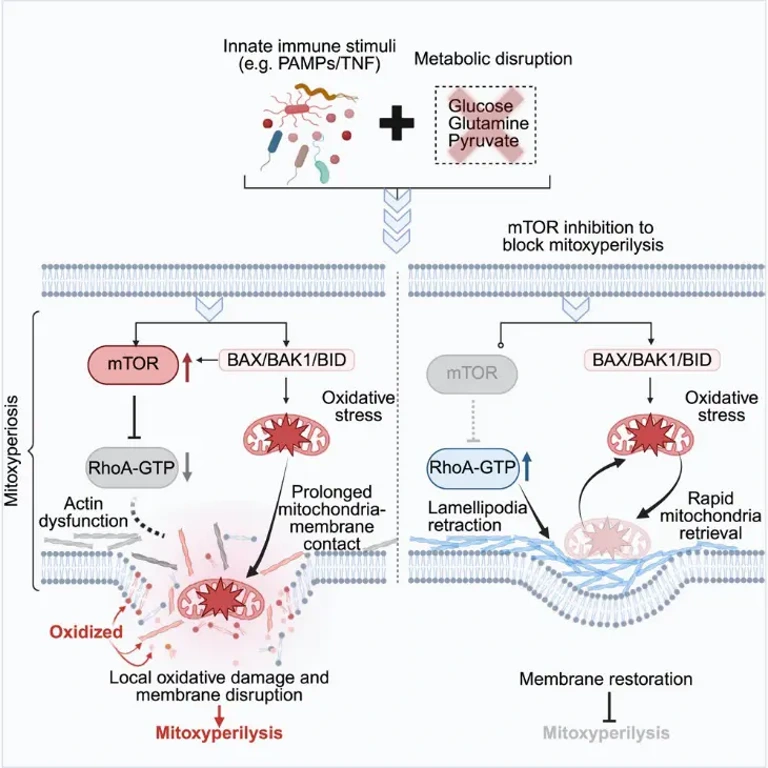
Mitochondria-Plasma Membrane Continuous Contact Mediates Local Oxidative Damage (Mitoxyperiosis)
Subcellular localization of oxidative stress is key to understanding the mechanisms of cell death.
-
Oxidative stress is localized to mitochondria: fluorescent spots highly co-localized with the mitochondrial probe, but not with the lysosomal probes, suggesting that IIAMD-induced oxidative stress mainly occurs in mitochondria;
-
Mitochondria maintain continuous contact with the plasma membrane: After LPS+CS treatment, mitochondria remained localized near the plasma membrane for an extended period (contact time > 20 min), while the contact time was shorter (<4 min) with LPS or CS treatment alone; furthermore, plasma membrane degradation and rupture subsequently occurred at the mitochondria-cell membrane contact sites;
-
Local oxidative damage drives membrane rupture: 4-hydroxynonenal (4-HNE, a lipid peroxidation marker) accumulated at the mitochondria-plasma membrane contact sites, and NAC treatment significantly reduced oxidative damage at this site; lipid peroxidation probe detection showed that lipid peroxidation levels gradually increased at the contact site before plasma membrane rupture.
mTORC2 Promotes Mitoxyperiosis by Inhibiting Cytoskeletal Activity
To screen for regulatory molecules of mitoxyperiosis, the team conducted a small molecule compound screen and found the following:
-
mTOR inhibitors have a significant protective effect: After screening 2050 compounds, it was found that the top 10 protective compounds included 3 mTOR kinase inhibitors; the mTOR dual inhibitor Torin-1 dose-dependently inhibited LPS+CS-induced cell death and was also effective in human macrophages (huMACs);
-
Dependence on mTORC2 rather than mTORC1: Knocking down the mTORC2 regulatory factor Rictor significantly inhibited cell death, while knocking down the mTORC1 regulatory factor Rptor or using the mTORC1 inhibitor rapamycin had no protective effect, demonstrating that mTORC2 is the key regulatory factor;
-
mTORC2 acts independently of oxidative stress: Torin-1 treatment did not affect intracellular GSH levels and oxidative stress intensity, but it prolonged the PI-negative time (maintaining membrane integrity); NAC treatment did not affect mTOR activation, suggesting that the two are independent regulatory pathways;
-
Molecular mechanism: mTORC2 promotes sustained contact between mitochondria and the cell membrane by inhibiting RhoA activity, reducing actin polymerization and membrane dynamics; Torin-1 treatment can restore RhoA activity, promote lamellipodia formation, move mitochondria away from the cell membrane, and reduce sustained contact and local oxidative damage; using the cytoskeletal inhibitor cytochalasin D (CyD) reversed the protective effect of Torin-1.
Core Differences Between Mitoxyperiosis and Known Cell Death Mechanisms
To clarify the uniqueness of mitoxyperiosis, the team compared its key characteristics with those of apoptosis, pyroptosis, necroptosis, and ferroptosis:
-
Differences in mitochondrial function: In mitoxyperiosis, the mitochondrial membrane potential (ΔΨm) gradually decreases, while in apoptosis (staurosporine-induced) and pyroptosis (LPS + nigericin-induced), ΔΨm collapses rapidly;
-
Differences in GSH levels: GSH levels significantly decrease in mitoxyperiosis, while there is no significant change in GSH levels in necroptosis (TSZ-induced) and pyroptosis;
-
Differences in mTOR dependence: Torin-1 can inhibit mitoxyperiosis, but has no protective effect on other forms of cell death.
In Vivo Validation: mTOR Inhibitors Alleviate IIAMD-Induced Inflammatory Damage and Tumor Necrosis
To verify the in vivo physiological significance of mitoxyperiosis, the team conducted two key experiments:
-
Alleviation of LPS + fasting-induced mouse death: Pre-injection of Torin-1 significantly increased the survival rate of "fasting + LPS" mice and reduced serum LDH and transaminase levels;
-
Regulation of tumor necrosis: In B16 tumor cells, LPS + CS can induce mitoxyperiosis, and knockdown of Rictor or Torin-1 treatment can inhibit cell death; in in vivo experiments, "intratumoral injection of LPS + fasting" can induce tumor necrosis, while Torin-1 treatment can reduce the necrotic area, and mTOR activity is increased and RhoA activity is decreased in tumor tissue, consistent with the in vitro mechanism.
This study is the first to report that mitochondria mediate local oxidative damage through sustained contact with the cell membrane, expanding the understanding of the mechanisms of mitochondrial involvement in cell death. mTORC2 inhibitors can alleviate inflammation and tumor necrosis induced by IIAMD, providing a new target for the treatment of infectious shock, tumor-related inflammation, and other diseases.
Cell-based assays can be used to measure the number of live cells, dead cells, and cells undergoing apoptosis, autophagy or oxidative stress in cell cultures. Our portfolio of cell-based assays uses luminescence or fluorescence detection to monitor various cellular events, helping you build a complete picture of how and why cells die.
Find out more about the different types of cell-based assays available.
Reference:
- Wang, Yaqiu, et al. "Innate immune and metabolic signals induce mitochondria-dependent membrane lysis via mitoxyperiosis." Cell 188.25 (2025): 7155-7174.
Your email address will not be published. Required fields are marked *