N6-methyladenosine Modification Regulates Iron Death Through Hepatic Stellate Cell Autophagy Signaling Pathway
Iron death is a recently discovered non-apoptotic form of cell death, which is characterized by iron-dependent lipid peroxidation. However, the exact underlying mechanism is still poorly understood. Researchers reported that the total level of N6-methyladenosine (m6A) modification increased significantly after exposure to iron death-inducing compounds due to the up-regulation of the methylase METTL4 and the down-regulation of the demethylase FTO.
Interestingly, RNA-seq showed that m6A modification seems to trigger autophagy activation by stabilizing BECN1 mRNA, which may be a potential mechanism for m6A modification to enhance HSC ferroptosis. Importantly, YTHDF1 has been identified as the key m6A reading protein for BECN1 mRNA stability, and the knockdown of YTHDF1 can prevent the HSC iron death induced by BECN1 plasmid. It is worth noting that YTHDF1 promotes the stability of BECN1 mRNA and autophagy activation by recognizing the m6A binding site in the coding region of BECN1.
In mice, erastin treatment reduces liver fibrosis by inducing HSC iron death. The specific inhibition of m6A-modified HSC can attenuate the iron death of HSC in mice with hepatic fibrosis induced by Ilastine. In addition, the researchers retrospectively analyzed the effect of sorafenib on HSC iron death and m6A modification in patients with advanced hepatocellular carcinoma (HCC) fibrosis who received sorafenib monotherapy. Interestingly, up-regulation of m6A modification, autophagy activation, and induction of iron death occur in human hematopoietic stem cells. Overall, these findings reveal new signaling pathways and molecular mechanisms of iron death, and also identify m6A modification-dependent iron death as a potential target for the treatment of liver fibrosis.
Liver fibrosis is a complex physiological and pathological condition, which is related to the healing mechanism. The deposition of extracellular matrix is the core event of liver fibrosis, and the myofibroblast matrix is mainly produced by activated HSC. Therefore, elimination of hematopoietic stem cells is the main treatment strategy for the development of anti-fibrosis treatments. Researchers have previously reported that the pathological and physiological conditions of liver fibrosis can be alleviated by triggering apoptosis, aging, necrosis, activation of adipocyte phenotypes, and preventing the proliferation, contraction, glycolysis, and pericytes of hematopoietic stem cells.
Interestingly, in recent studies, iron death is considered to be a new and effective method to eliminate hematopoietic stem cells. The researchers found that the RNA-binding proteins ELAVL1 and ZFP36 can trigger HSC ferroptosis by regulating the autophagy pathway to alleviate liver fibrosis. In addition, they also found that the mitochondrial iron metabolism pathway may regulate HSC iron death through the BRD7/P53/SLC25A28 axis. The purpose of this study is to evaluate the underlying mechanism of iron death and its role in the suppression of liver fibrosis.
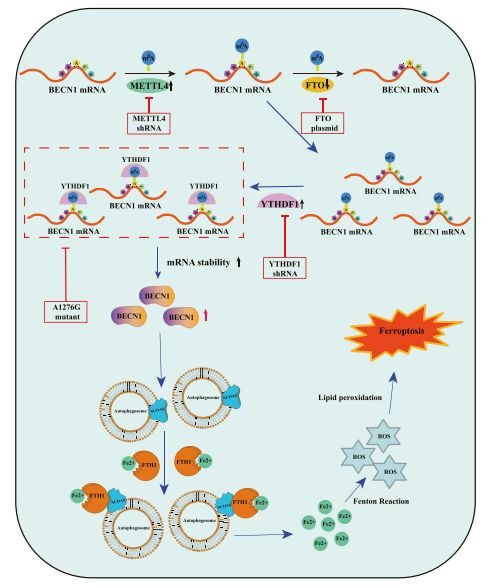
Different from the typical morphological characteristics of apoptosis, necrosis, necrosis, pyrolysis and aging, iron death is a recently discovered form of programmed cell death. Iron-dependent lipid peroxidation, loss of glutathione peroxidase 4 (GPX4) and concentration of mitochondrial membrane density are the main features of iron death. According to reports, system Xc inhibition (such as sulfapyridine, olafenib, erastin), GPX4 inhibition (such as FIN56, atetamine, RSL3, FINO2) and physiological conditions (such as cystine deprivation, amino acid starvation, high extracellular gluten) Acid) can induce iron death. Importantly, the mutual interference between iron death and autophagy has received more and more attention, and autophagy is a targeted way to regulate the sensitivity of cells to iron death. Although the transcriptional regulation of autophagy signaling pathway in iron death has been elucidated, the role of post-transcriptional regulation in iron death is still poorly understood.
m6A RNA modification is the most abundant post-transcriptional mechanism in eukaryotic messenger RNA (mrna). It is a reversible process controlled by methyltransferase, demethylase, and m6A binding protein. The methyltransferase complex catalyzes the methylation of m6A mRNA, which includes WT1-related protein (WTAP), methyltransferase like 3 (metttl3), metttl4 and metttl14. AlkB homolog 5 (ALKBH5) and obesity-associated protein (FTO) act as demethylases to remove m6A modifications from RNA, thereby maintaining the dynamic balance of m6A modifications. In addition, m6A binding proteins include heterogeneous ribonucleoprotein A2/B1 (HNRNPA2B1), insulin-like growth factor 2 mRNA binding proteins (IGF2BPs), YTH domain-containing protein 1/2 (YTHDC1/2) and YTH domain family 1 / 2/3 (YTHDF1/2/3) binds to the m6A motif to affect RNA stability or function. Interestingly, exploring the post-transcriptional regulation of m6a-mediated stellate cell iron death may provide therapeutic targets and effective diagnostic indicators for liver fibrosis.
The study analyzed a new signaling pathway and molecular mechanism of iron death in liver fibrosis. The study found that the m6A reader YTHDF1 promotes the stability of BECN1 mRNA by identifying the m6A binding site, thereby triggering autophagy activation and ultimately leading to the death of HSC iron. The authors pointed out that m6A modification may be a new and key post-transcriptional regulator of iron death in liver fibrosis.
Reference:
- Min Shen et al. N6-methyladenosine modification regulates ferroptosis through autophagy signaling pathway in hepatic stellate cells. Redox Biol 2021 Sep 26;47:102151. doi: 10.1016/j.redox.2021.102151.
Your email address will not be published. Required fields are marked *